


What can we say generally about the effect of complexity, such as geometrical complexity and multi-metallicity, on catalytic performance? Can we expect further, equally large improvements when growing more dendritic structures or adding yet more metals? Given the novel dielectric-matrix based structures, can the semi-metallic compounds’ influence, such as that of carbon matrixes,[3] which have been used for catalytic applications since long before the advent of nanotechnology, be understood as the fact of carbon or a semiconductor behaving like “half a metal”? What ways of adding metals take most advantage of or enhance the fractal structure in order to obtain interesting new catalysts?
Adding another metal to a catalyst that is already optimized for specific reactions will likely destroy the optimization. There are at least two more metal-to-metal atomic ratios when adding a single metal to a bimetallic compound (plus relative locations etc.). The fact that re-optimization occurs in a parameter space with a higher dimensionality, while the previous optimization is a maximum constrained on a lower dimensional subspace, already explains why properties can likely be improved.
How to understand this? Well, start considering being already in the higher dimensional space, for example on a mountain in 3D space. Assume for simplicity that there is one peak, the maximum. It is somewhere in this 3D space. The previous optimization was done in a 2D subspace. The previous 2D subspace may be some random cut through the 3D space. The probability that the previous 2D subspace included the location of the 3D peak is basically zero! The other way around: If you are constrained to be on a lower dimensional subspace such as a walk way on the mountain, even if you are on the highest point that the path allows, you must leave the path so you can reach the true peak.
However, that properties can improve does not explain why they are often already improved by adding a second or third metal, even before re-optimization! A catalyst is an agent that (or who) accomplishes a certain task, thereby not only often speeding up a reaction but enabling it. The catalyst, just like in the social realm, does something. It does take part in the reaction although by definition, a catalyst reemerges unchanged after having undergone the crucial processes.
A modern compound catalyst completes its tasks in a ‘modular’ fashion, much like any complex catalytic agent (say in the social context). There are a multitude of different sub tasks such as proton absorption, electron transfer, removal of poisoning intermediates, etc., and they are accomplished by different parts. For example, two micrometer long, front-Pt-back-AgAu-alloy nano tubes can be propelled at 150 micrometer/s (Pt ahead) inside H2O2 solution. The Pt surface takes the H2O2 apart (H2O2 --> 2H+ + 2e- + O2), hands the protons and electrons to the alloy, and then the alloy reassembles them differently (4H+ + 4e- + 2O2 --> 2H2O + O2). The locomotion can involve gas expulsion (with nano tubes) as well as proton gradients and dipole fields.[4,5]
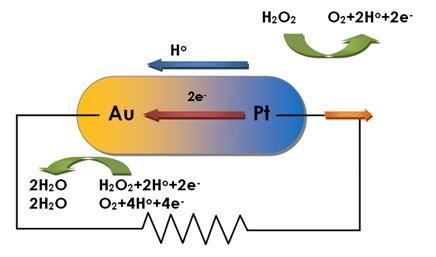
Considering synergy of required tasks such as electron exchange through a metal-metal interface, proton transfer through Pd, restricting access to reaction sites to desired molecules via the matrix, and others, makes understood that going bi- or tri-metallic often leads to huge enhancements in catalytic strength. The reaction finds a more convenient way in the more complex environment even before we re-optimize parameters.
We cannot expect similar by increasing from four to five different metals, if this increases the complexity far beyond the complexity of the reaction that is to be catalyzed.
On one hand, the magic is demystified: We understand that the further addition of a metal should best add the capability for another task, for example adding Pd [6] (or even just Pt [7,8]) will likely add hydrogen absorbance and transfer capability because Pd absorbs hydrogen and is thus extensively used in hydrogen sensing, [9] is quite generally very catalytic [10] and extensively used to catalyze low temperature reduction of pollutant gases [11] and organic reactions.[12] On the other hand, we cannot know what is possible in a new parameter space, and any new addition that adds complexity may enable new tasks.
Merely using an alloy combining ten different metals will not work. “The more mixed the better” (for complexity or social "diversity") is certainly wrong. An atomic alloy where every Pd atom is surrounded by Au and vice versa, does neither provide good hydrogen absorption, nor does it provide metal to metal interfaces; there are no electron depletion zones on the nanometer scale. Complexity improves performance only if it is “true complexity” that, for example, provides the versatility of required tasks (true diversity).
Seeking complexity can be counter-intuitive and unnatural. Especially in catalysis and before optimization, we aim for broad distributions in crystal sizes and imperfect shapes, rather than for the usually advertised narrow control over sizes and aesthetically appealing microscopy images. A ‘smearing’ of complexity over different scales (fractality) is generally justified:
A huge specific surface area on the nanoscale is useless if all nano pores are only accessible through other nano pores. In a scale invariant network, small pores are accessible through a chain of increasingly larger pores.
The broad pore size distribution of a nano-sponge extends over four orders of magnitude from 10-8 to 10-4 meters. The dentritic structure of a treelike catalyst enables electron transport on all scales, starting with the current that we want to draw from an electro-chemical device such as a fuel cell or supercapacitor, all the way down to the electron transport on the nano and atomic scale, where electron re-distributions offer a complex and dynamic environment so that electro-chemical reactions can find pathways that are otherwise closed to them. Also because of this desired ‘smearing of complexity’ is it worth to quantify and control the statistics of distributions while monitoring performance.
Catalytic strength may be explored with the help of well known benchmark reactions like the bleaching of RhodamineB and methanol electro-oxidation. Optimizing such reactions however cannot show whether the catalyst is complex enough for more complex reactions. No real world application needs to bleach RhodamineB fast!
The problem is that we cannot quantify “true complexity”. Scientists disagree on what complexity is, and this issue may never be settled. Complexity’s “magic” may well be necessarily related to that any true increase in complexity escapes previous ways of quantification. This brings us to the issue of emergent parameters that I will discuss the next time.
--------------------
This article continues the series “Adapting As Nano Approaches Biological Complexity: Witnessing Human-AI Integration Critically”, where the status of all this as suppressed information has been discussed. I allow myself to actually publish the most interesting and critical parts here (edited). If citing, please cite nevertheless [1] anyway in order to support the author.
The previous post was “Complexity And Optimization: Lost In Design Space”
The next post continues with
"Emergent Parameters and High Throughput Screening" before finally getting to
"Critique of Nanotech: The most dangerous Science least carefully done?"
and perhaps I also add
"A flexible, evolving approach to computing."
References:
[1] S. Vongehr et al., Adapting Nanotech Research as Nano-Micro Hybrids Approach Biological Complexity. J. Mater. Sci.&Tech. 32(5), 387-401 (2016) doi: 10.1016/j.jmst.2016.01.003
[2] C. J. Zhong, J. Luo, P. N. Njoki, D. Mott, B. Wanjala, R. Loukrakpam, S. Lim, L. Wang, B. Fang and Z. Xu, Energy Environ. Sci., 2008, 1, 454–466.
[3] V. Celorrio, M. G. Montes de Oca, D. Plana, R. Moliner, M. J. Lazaro and D. J. Fermin, Effect of Carbon Supports on Electrocatalytic Reactivity of Au-Pd Core-Shell Nanoparticles. J. Phys. Chem. C, 2012, 116, 6275-6282.
[4] W. F. Paxton, S. Sundararajan, T. E. Mallouk and A. Sen, Chemical Locomotion. Ang. Chem. Int. Ed., 2006, 45, 5420-5429.
[5] W. F. Paxton, A. Sen and T. E. Mallouk, Motility of Catalytic Nanoparticles through Self-Generated Forces. Chem. A Eur. J., 2005, 11, 6462-6470.
[6] J. F. Huang, S. Vongehr, S. C. Tang, H. M. Lu, J. C. Shen and X. K. Meng, Highly Catalytic Pd-Ag Bimetallic Dendrites. J. Phys. Chem. C, 2010, 114, 15005-15010.
[7] Y. Xia, J. Liu, W. Huang and Z. Li, Electrochemical fabrication of clean dendritic Au supported Pt clusters for electrocatalytic oxidation of formic acid. Electrochim. Acta., 2012, 70, 304-312.
[8] S. C. Tang, S. Vongehr, Z. Zheng and X. K. Meng, Co dendrite based bimetallic structures with nanoflake-built Pt covers and strong catalytic activity. J. Coll.&Interf. Sci., 2010, 351, 217-224.
[9] T. Shegai, P. Johansson, C. Langhammer and M. Kall, Directional Scattering and Hydrogen Sensing by Bimetallic Pd-Au Nanoantennas. Nano Lett., 2012, 12, 2464-2469.
[10] S. Uhm, H. J. Lee and J. Lee, Phys. Chem. Chem. Phys., 2009, 11, 9326–9336.
[11] H. Tanaka, M. Uenishi, M. Taniguchi, I. Tan, K. Narita, M. Kimura, K. Kaneko, Y. Nishihata and J. Mizuki, Catal. Today, 2006, 117, 321–328.
[12] S. U. Son, Y. Jang, J. Park, H. B. Na, H. M. Park, H. J. Yun, J. Lee and T. Hyeon, J. Am. Chem. Soc., 2004, 126, 5026–5027.



Comments