


Previous research has indicated that DR Cysteamine can prevent abnormal neuronal cell death caused by Huntington’s. Preclinical studies conducted by Drs. Sandrine Humbert and Frederic Saudou from the Curie Institute demonstrated in 2006 prevention of neuronal apoptosis through usage of cysteamine. Treatment with cysteamine is linked to increased levels of Brain derived neurotrophic factor (BDNF) which protects brain cells and promotes growth of new brain cells. Their study showed an increase in both brain and blood levels of BDNF, which allows this protein to be used as a biomarker. BDNF is known to be depressed in patients with Huntington’s. If proven efficacious and viable, treatment with DR Cysteamine could protect from the neuronal cell death by increasing levels of protective BDNF.
"We are encouraged by the existing studies that indicate cysteamine's potential as a neuroprotectant for treating Huntington's, and we believe that Raptor's formulation could offer a better tolerated drug for this patient population,” said geneticist and professor Dominique Bonneau, M.D., Ph.D. at CHU d'Angers. “We are confident that this trial will enhance our understanding of cysteamine's effects in Huntington's patients and we hope it may lead to a new treatment for this serious, debilitating disease."
The two year study, funded by the French Ministry of Health, will examine the effect of DR Cysteamine treatment in patients with diagnosed Huntington’s. Endpoints of the clinical trial will evaluate levels of BDNF and also observe patient symptoms according to the Unified Huntington’s Disease Rating Scale (UHDRS). The scale will examine motor, cognitive, and behavior functions of patients participating in the study.
Cysteamine is already approved by the FDA to treat nephropathic cystinosis, a rare genetic metabolic disorder affective lysosomal storage. Cystinosis can result in kidney failure and treatment with cysteamine can delay or prevent kidney transplant.
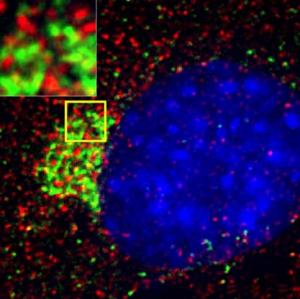
Photo: A montage of three images of single striatal neurons transfected with a disease-associated version of huntingtin, the protein that causes Huntington's disease. Nuclei of untransfected neurons are seen in the background (blue). The neuron in the center (yellow) contains an abnormal intracellular accumulation of huntingtin called an inclusion body (orange). Studies using an automated microscope and survival analysis demonstrated that neurons with disease-associated Huntingtins that form inclusion bodies survive longer than those that do not. Photo credit: Steven Finkbeiner, M.D., Ph.D, the Gladstone Institute of Neurological Disease and the University of California, San Francisco
Huntington’s disease is a rare genetic condition characterized by degenerative brain cell death. Gene IT15 codes for a protein, huntingtin, which has unknown activity. When this gene becomes mutated, it produces mutant huntingtin specifically acting in the striatum region of the brain, responsible for motor control, and results in abnormal cell death. Onset occurs between 35-50 years of age and results in
death 15-20 years after diagnosis due to fatal complications of neuronal degeneration most likely from choking, respiratory failure or infection.
The Huntington’sDisease Society of America (HDSA) classifies the disease into three stages of progression with early stages marked by changes in coordination, involuntary movements, and difficulty in comprehension. Uncontrolled movements become more distinct in middle stages of disease progression followed by diminished ability for speech, swallowing and daily activities. Late stage progression of Huntington’s is characterized by inability to complete daily activities. Inability to swallow may cause choking. Motor control is reduced further, as the patients can no long walk or talk. Huntington’s Disease is a dominant genetic trait in which a child has a 50% change of developing the disease if either parent was diagnosed with Huntington’s. There are currently 30,000 people
living with Huntington’s Disease in the United States and 150,000 others who live with the possibility of a future diagnosis.
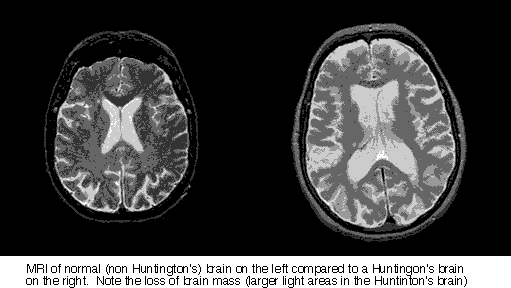
Photo Credit: Macalester College
There is currently no treatment for Huntington’s Disease. Medication is prescribed for management of mood and motor control, but no treatment exists for the underlying cause. There are currently a number of investigative studies exploring many avenues of treatment.
If proven to be clinically effect, DR Cysteamine may be used as an effective treatment for those diagnosed with Huntington’s and for those with the chance of developing the disease. If demonstrated viable, a treatment for Huntington’s could save thousands of lives cut short by this debilitating condition and could lead us one step further into uncovering the mystery of disease and the influences of our genetics.
References:
Cystamine and cysteamine increase brain levels of BDNF in Huntington disease via HSJ1b and transglutaminase J. Clin. Invest. Maria Borrell-Pagès, et al. 116:1410 doi:10.1172/JCI27607National Institutes of Health- Huntington's Disease
Huntington's Disease Society of America



Comments