


Some people age faster than others and a new study hopes to be able to measure the transcriptome, gene expression, and create a true way to read biological clocks. This study was with the model organism Caenorhabditis elegans, and worms are not little people, but for the last four decades it has become vital for studying DNA and genomes.
Aging clocks such as Horvath's epigenetic clock are limited because they are based on the pattern of methylations, small chemical groups that attach to DNA and change with age. Using the transcriptome, the new clock takes into consideration the set of genes that are read from DNA (messenger RNA) to make proteins for the cell but the transcriptome was considered too complex to indicate age. Sometimes genes transcribe a particularly large amount of mRNA, sometimes less.
It has not been possible to develop precise aging clocks based on gene activity. Meyer and Schumacher's new approach uses a mathematical trick to eliminate the differences in gene activity. The proposed binarized transcriptome aging clock divides genes into two groups - 'on' or 'off' - thus minimizing high variation. This makes aging predictable from the transcriptome.
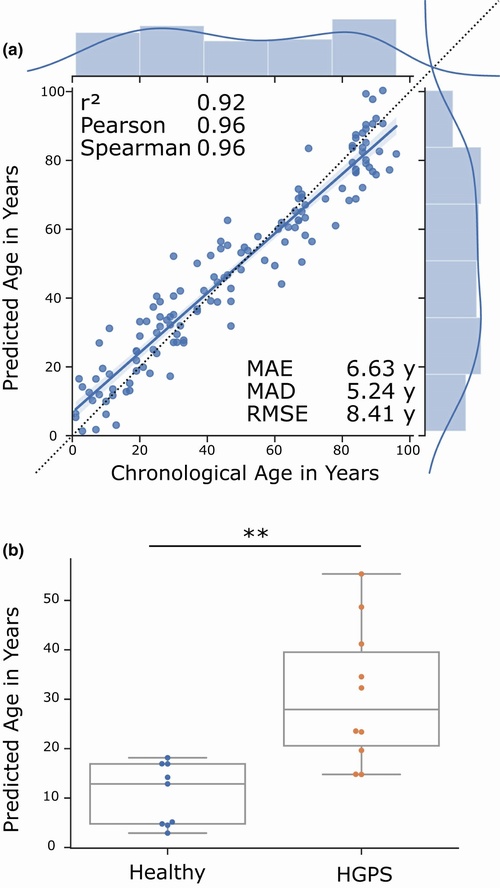
Transcriptomic human aging clock. (a) Results of the age prediction computed by cross‐validation on human fibroblast gene expression data. The x‐axis shows the chronological age in years. The y‐axis shows the predicted age computed by an elastic net regression on binarized gene expression data. Every blue dot displays one RNA‐seq sample. The regression line with the 95% confidence interval is shown in blue, and the dotted line shows the perfect linear correlation. The distribution of the data is shown on the side of the plot. r2 = coefficient of determination, Pearson = Pearson correlation, Spearman = Spearman correlation, MAE = mean absolute error in years, MAD = median absolute deviation in years, RMSE = root‐mean‐square‐error in years. Data from GSE113957. (b) Box plots of age predictions of samples from Hutchinson‐Gilford progeria syndrome patients (red) and predictions of age‐matched healthy controls (blue) by the elastic net regression of binarized gene expression data. Progeria samples are predicted to be significantly older than age‐matched healthy controls. Data from GSE113957. **p ≤ 0.01, calculated by an independent two‐sided t test. Table S3 contains more detailed statistics
BiT age is based exclusively on approximately 1,000 different transcriptomes of C. elegans, for which the lifespan is precisely known. Model organisms such as the nematode provide a controllable view of the aging process, allowing biomarkers to be discovered and the effects of external influences such as UV radiation or nutrition on longevity to be studied.
The new aging clock allows researchers to accurately predict the pro- and anti-aging effects of gene variants and various external factors in the nematode at a young age. The aging clock also showed that genes of the immune response as well as signalling in neurons are significant for the aging process. 'BiT age can also be applied to predict human age quickly and with very high accuracy. Measuring biological age is important to determine the influence of environment, diet or therapies on the aging process and the development of age-related diseases. This clock could therefore find wide application in aging research. Since BiT age is based purely on gene activity, it can basically be applied to any organism,' Schumacher explained.



Comments