


New research shows that cystic fibrosis (CF), a life-shortening, inherited condition that affects about 30,000 Americans, causes a specific defect in this process, reducing the ability to clear particles and germs out of the airway.
In CF, mucus abnormalities are linked to serious health problems, including airway obstruction, difficulty breathing, and increased susceptibility to lung infection. However, whether the mucus abnormality is a primary defect in cystic fibrosis, or is a secondary consequence of CF lung disease, has been an unanswered question.
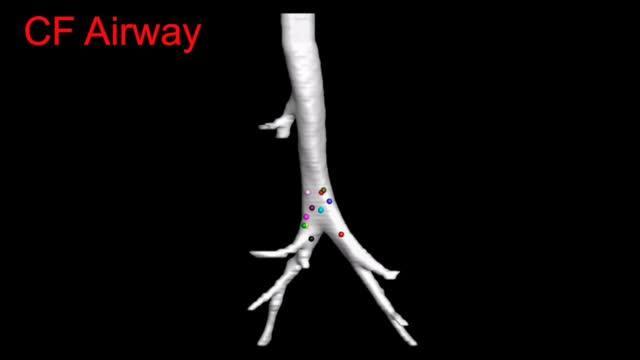
3-D image reconstruction from time-lapse CT-scans of a CF pig lung. Credit: Maged Awadalla, Mark Hoegger and the University of Iowa.
Using a pig model of cystic fibrosis, which more closely mimics human disease than the traditional mouse experimental models, the UI team found that the mucus defect is present at birth and is a primary cause of CF lung disease.
The team discovered that mucus emerges from specialized glands below the airway surface in long strands. In normal, non-cystic fibrosis airways, these strands detach and are swept up to the throat carrying any trapped particles with them. However, in
cystic fibrosis
airways these strands don't detach properly from the gland, which allows mucus and trapped particles to accumulate on the airway surface (see video 1).
"If you can't clear mucus out of the airway properly, the airway can get plugged up making it very difficult to breathe, and that's what we see in people with CF," explains Tony Fischer, MD, PhD, study author and pediatric pulmonary fellow with UI Children's Hospital.
"Now that we know more about what we are up against in CF, we can direct our research," adds Mark Hoegger, first author of the study and a student in the UI Medical Scientist Training Program. "We can try to figure out ways to prevent these mucus strands from forming to begin with, and we can figure out ways to detach tethered mucus that has already formed."
Most of the research involved high-tech imaging techniques using confocal microscopes, CT scanners, and 3D image analysis, but a key experiment was possible only by using decidedly simpler technology -- an iPhone and a $5, stop-motion-capture app -- to visualize the movement of particles across airway surfaces (see video 2).
Under normal conditions, movement of particles across CF and non-CF airways looked similar. However, when the airways were made to produce excess mucus, particles in the CF airway would sometimes get stuck and remain attached to the airway. To find out why, Hoegger used fluorescent nanospheres to visualize static mucus and found that in CF airways there were strands and blobs of mucus attached to the airway surface specifically at the mucus gland openings.
Viewing the videos showing the mucus defect created mixed emotions for the researchers.
"When you look at these pictures and videos you understand very quickly what is going on, and as scientists we were very excited about these discoveries," Hoegger says. "But after seeing that the mucus is not properly detaching from the airways, we reflected on the challenges that this creates for people with CF."
The new findings also directly link the mucus defect to the loss of function of the CFTR protein – the underlying genetic cause of CF. The research team was able to recreate the mucus defect in non-CF airways by shutting down chloride and bicarbonate ion secretion in the mucus glands. The CFTR protein normally controls this ion movement, so the findings directly link loss of CFTR function to the impaired mucus clearance in CF.
The newly discovered CF mucus abnormality may also play a role in problems caused by the disease in other organs, like the GI tract and the pancreas, and the finding may also have implications for other airway diseases with abnormal mucus and mucociliary transport such as asthma and COPD.



Comments